Fatores críticos de sucesso para o seu estudo clínico

Os estudos clínicos em Portugal
Os ensaios clínicos de medicamentos de uso humano e os estudos clínicos com intervenção de dispositivos médicos são uma etapa importante no processo de inovação e desenvolvimento, onde existe o rigoroso desenho de um protocolo a fim de apurar a eficácia / desempenho (no caso dos dispositivos médicos) e segurança em seres humanos.
Nos últimos anos, Portugal tem vindo a evoluir na sua legislação, a acompanhar as diretrizes europeias, e a procurar criar iniciativas e organismos que visam a agilização e o rigor ético dos processos de investigação clínica. Graças a este contexto regulamentar mais favorável e empenho de diversos quadrantes da sociedade, tem-se vindo a registar uma evolução positiva na realização de estudos clínicos em Portugal. Poderá verificar em maior detalhe esta evolução na secção Portugal em Números.
A condução dos ensaios clínicos na União Europeia (UE) foi recentemente objeto de uma alteração profunda quando o Regulamento dos Ensaios Clínicos (Regulamento (UE) n.º 536/2014) entrou em vigor a 31 de janeiro de 2022. Este regulamento harmoniza os processos de submissão, avaliação e supervisão dos ensaios clínicos na UE através do Sistema de Informação de Ensaios Clínicos (CTIS) que é o ponto de entrada único para a submissão de informações relativas aos ensaios clínicos na UE e no Espaço Económico Europeu (EEE). O CTIS funciona como um websitepúblico que oferece ao público em geral, informações detalhadas sobre os ensaios clínicos conduzidos na UE e no EEE desde o momento em que os respetivos pedidos são aprovados no CTIS. Além disso o CTIS tem um ambiente de trabalho destinado aos promotores de ensaios clínicos ou seus representantes e um ambiente de trabalho destinado às autoridades dos Estados-membros da UE e dos países do EEE e à Comissão Europeia.
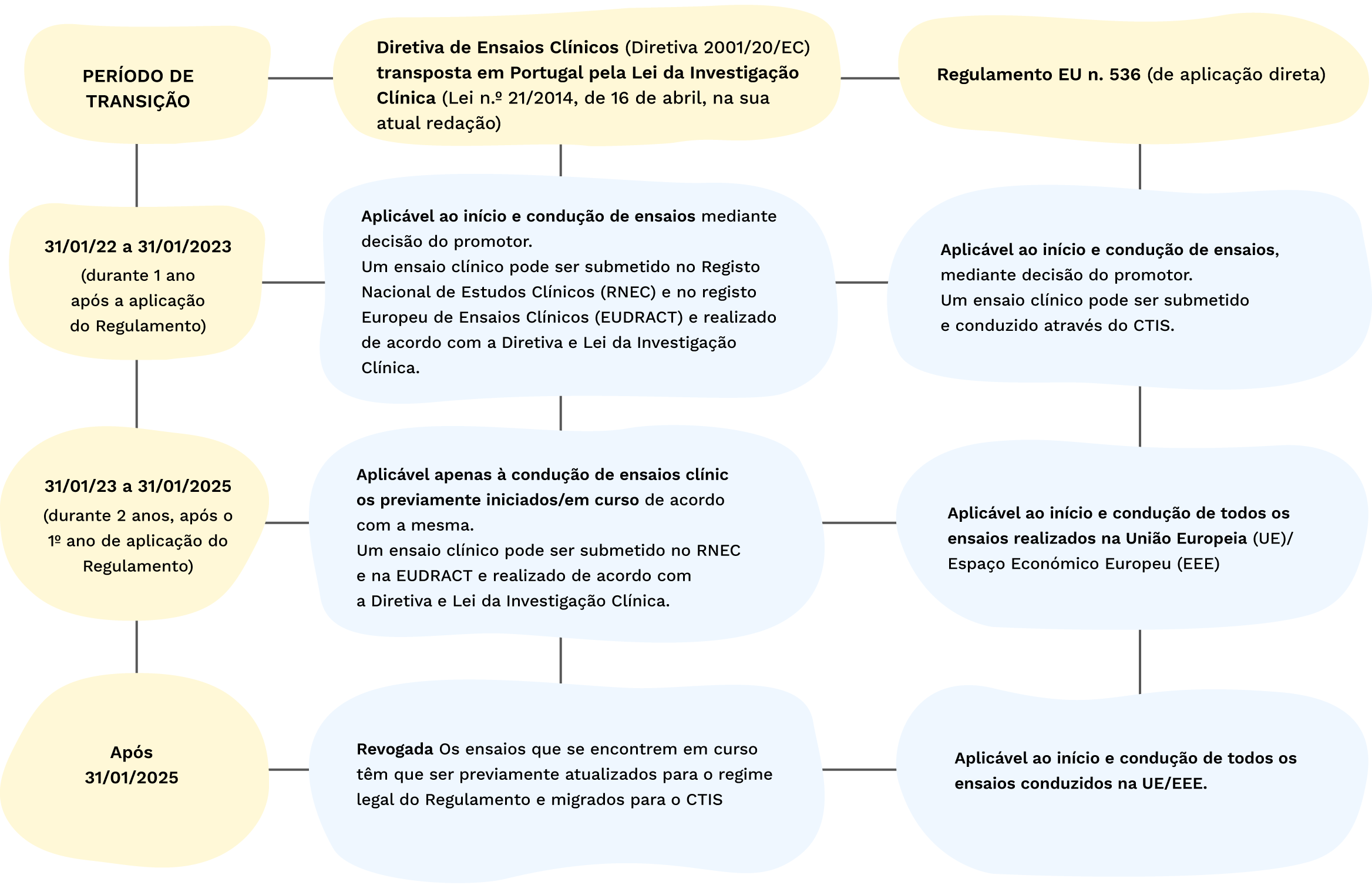
O Regulamento dos Ensaios Clínicos prevê um período de transição para o CTIS de três anos:
- De 31 de janeiro de 2022 a 30 de janeiro de 2023, os promotores de ensaios clínicos puderam optar por submeter os seus pedidos de ensaios clínicos ao abrigo da Diretiva dos ensaios clínicos (Diretiva 2001/20/CE) através da submissão de processos a nível nacional, ou ao abrigo do Regulamento dos Ensaios Clínicos através do CTIS;
- A partir de 31 de janeiro de 2023, todos os novos pedidos de ensaios clínicos na UE e no EEE são submetidos ao abrigo do Regulamento dos Ensaios Clínicos através do CTIS;
- Até 30 de janeiro de 2025, os ensaios aprovados ao abrigo da Diretiva dos ensaios clínicos que ainda estejam em curso terão de ser transferidos para o procedimento ao abrigo do Regulamento dos Ensaios Clínicos e para o CTIS.
Para além desta regulamentação, em Portugal deverá ser tida em conta ainda a legislação nacional estabelecida pela Lei n.º 21/2014, de 16 de abril (Lei de Investigação Clínica), na sua atual redação pela Lei n.º 73/2015 de 27 de julho e Lei nº Lei n.º 49/2018, de 14 de agosto. Para além desta legislação nacional, os medicamentos de uso humano e dispositivos médicos são regidos por regulamentação adicional. Poderá consultar a informação regulamentar mais relevante sobre medicamentos de uso humano e dispositivos médicos na tabela abaixo:
Regulamentação
Ensaios clínicos de medicamentos de uso humano
-
Autoridade competente nacional
- INFARMED, I.P.
-
Comissão de ética competente
- CEIC
- Canal de submissão
- Legislação Europeia
- Legislação Nacional
- Legislação Complementar
Estudos clínicos com intervenção de dispositivos médicos
-
Autoridade competente nacional
- INFARMED, I.P.
-
Comissão de ética competente
- CEIC
- Canal de submissão
- Legislação Europeia
- Legislação Nacional
- Legislação Complementar
Entidades envolvidas
De acordo com a legislação em vigor, o INFARMED, I.P. é a autoridade competente em matéria de ensaios clínicos que envolvam medicamentos de uso humano e estudos clínicos com intervenção de dispositivos médicos ou de produtos cosméticos ou de higiene corporal. Todos os restantes estudos clínicos são da responsabilidade da Comissão de Ética para a Investigação Clínica (CEIC).
Para que um ensaio clínico seja realizado em Portugal, é necessário que seja dado um parecer favorável pela Comissão de Ética competente em Portugal, a CEIC, e seja dada autorização pelo Conselho Diretivo do INFARMED, I.P..
Para a implementação de um estudo clínico, incluindo um ensaio clínico, o promotor recorre a Centros de Investigação Clínica, inseridos em hospitais ou instituições de Saúde ou de Investigação com recursos materiais, técnicos e humanos, com uma Comissão de Ética Clínica e equipas de médicos especializados, coordenadores de estudos clínicos, enfermeiros e farmacêuticos que irão garantir o cumprimento das boas práticas clínicas e dos princípios éticos que as mesmas salvaguardam.
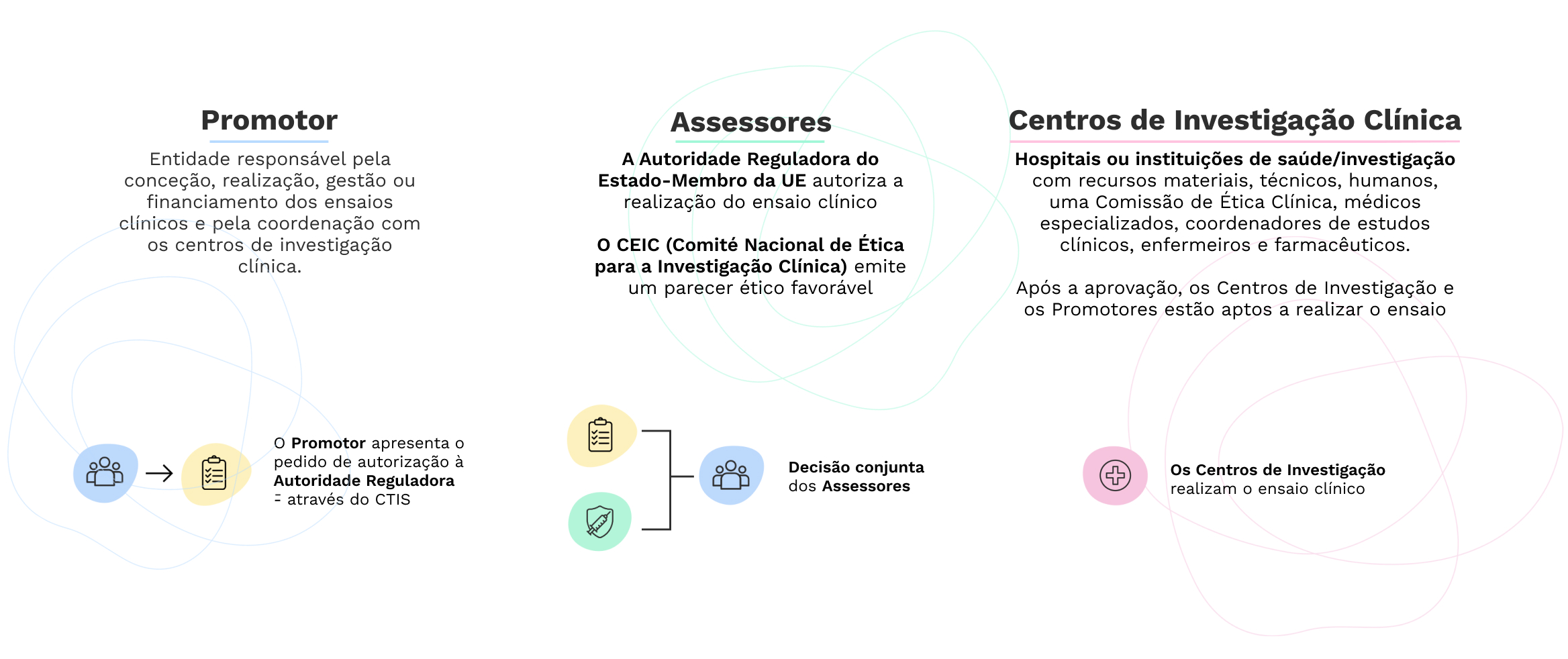
Processo de submissão e implementação de ensaios clínicos com medicamentos ao abrigo do Novo Regulamento 536/2014
O Regulamento de ensaios clínicos, Regulamento EU n. 536 entrou em vigor a 31 de janeiro de 2022.
Entre outros aspetos, o Regulamento EU de ensaios clínicos altera o sistema de submissão, avaliação e supervisão dos ensaios clínicos na União Europeia (EU) e nos países do Espaço Económico Europeu (EEE), que passa a efetuar-se através de um Portal (EU Portal and Database, EUPD) que constitui o ponto de acesso a um sistema europeu integrado de informação sobre ensaios clínicos (Clinical Trials Information System, CTIS) que entrou em funcionamento na mesma data.
O CTIS tornar-se assim o ponto único para submissões, registo de informação e emissão de autorizações de pedidos de ensaios clínicos, de acesso restrito, dando suporte operacional aos processos dos utilizadores (tanto autoridades competentes nacionais (ACN) e Comissões de Ética (CE) dos Estados-membros (EM) como promotores).
Esquema e Tabela do período de transição:
O CTIS também pretende tornar a informação sobre ensaios clínicos na Europa mais acessível e transparente. Está assim sujeito a regras de transparência e interage com 5 bases de dados da Agência Europeia do Medicamento (EMA, na sigla inglesa) permitindo aos utilizadores pesquisarem informação sobre as organizações envolvidas e sobre os medicamentos. Inclui igualmente uma área aberta para informação ao cidadão e ao público em geral.
O Regulamento revoga a Diretiva de ensaios clínicos de forma faseada, por aplicação de um período de transição.
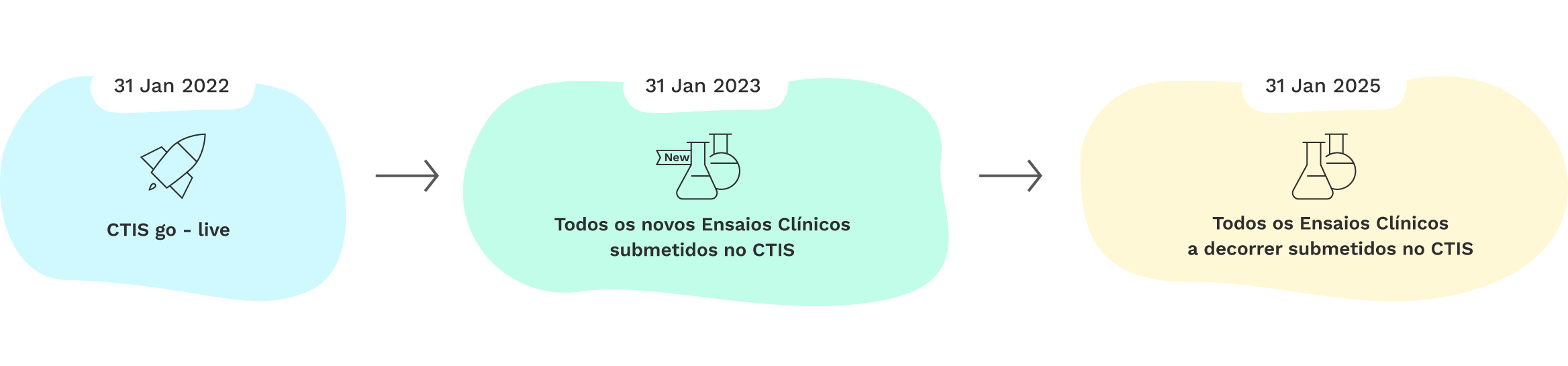
Tabela Período de Transição
.
Preparação para entrada em vigor do Regulamento
- Atender ao período de transição no planeamento do regime legal que pretendem aplicar ou para o qual terão que transitar os seus ensaios, atualmente em curso e/ou em preparação, atendendo à calendarização das atividades regulamentares durante o ciclo de vida do ensaio.
- Ajustar os seus processos e a gestão dos direitos de acesso/edição/submissão dos utilizadores de CTIS à forma como pretendem planear e gerir os seus ensaios de acordo com o Regulamento e através do CTIS.
- Obter informação e formação sobre o CTIS para aplicação do novo Sistema, que é apoiada por extensos programas preparados pela Agência Europeia de Medicamentos (EMA na sigla em inglês) responsável pela sua manutenção. Estes programas são tanto de caracter geral como dirigidos aos diferentes tipos de entidades envolvidas na realização, avaliação e supervisão de ensaios clínicos. Assim, é muito relevante que tanto os promotores comerciais como os não comerciais/académicos consultem os extensos e abrangentes conteúdos formativos que o site da EMA disponibiliza.
- Guia de consulta dos documentos publicados pela EMA mais relevantes para Promotores e Investigadores/promotores académicos, que pretendam implementar o Regulamento de Ensaios Clínicos 536/2014 de 16 de abril nos seus processos e programas de formação - Download do Guia.
- Do ponto de vista operacional, haverá que atender a aspetos previamente à submissão de um pedido para início de um ensaio clínico no CTIS, nomeadamente:
- Garantir que os dados relevantes estão registados nas bases de dados e nos sistemas da EMA com que o CTIS interage, nomeadamente o EMA Account Management, (para registo de utilizadores), o OMS (Organisation Management Service, para registo/pesquisa de entidades) e o xEVMPD (Extended EudraVigilance medicinal product dictionary, na sigla inglesa, para registo/pesquisa de medicamentos). O estado dos dados registados no OMS, em caso de dúvida da parte do promotor, pode ser verificado através do SPOR (Substance, product, organisation and referential portal) da EMA.
- Criar uma conta da EMA necessária para acesso à área de trabalho restrita “dos promotores”.
Nota: Informação sobre como registar um administrador do promotor está disponível no Programa online de formação modular sobre o CTIS da EMA CTIS online modular training programme, em particular no módulos 3 e 19 e nos guias passo-a-passo e tutoriais de vídeo ( CTIS Highlightsnewsletter). O registo para administradores CTIS (high-level) está aberto desde setembro de 2021.
- Ao ser considerado o registo de utilizador no CTIS as organizações devem ponderar se pretendem assumir uma abordagem centrada:
- na Organização ou
- no Ensaio Clínico
(organization Centric vs. CT-centric approaches)
Nota: Informação sobre como registar um administrador do promotor está disponível no modulo 7 do Programa online de formação modular sobre o CTIS da EMA CTIS online modular training programme
Para mais informações consultar os sites:
Procedimento expedito de avaliação de Ensaios Clínicos com medicamentos para a COVID-19 – 01/02/2022 – 31/01/2024
No contexto da estratégia da União Europeia para resposta à necessidade de investigação de tratamentos para a COVID-19, a Comissão Europeia lançou uma Joint Action (JA), no âmbito da EU4Health para avaliação coordenada e acelerada de ensaios clinicos multinacionais tendo a COVID19 como indicação tratamento. Este procedimento de avaliação, ao abrigo do novo Regulamento de ensaios clínicos terá início a partir de fevereiro de 2022, contando com a participação de Portugal.
A fim de transmitir aos potenciais promotores de ensaios clínicos informações sobre esta ação conjunta, foi organizado pelo Coordenador da JA e pelo Steering Group, uma sessão de informação dirigida às partes interessadas quando pretendam realizar ensaios multinacionais.
Vídeo da sessão de divulgação desta ação disponivel em:
Estudos clínicos com intervenção de dispositivos médicos